Клиничен обзор върху идиопатичните интерстициални пневмонии
Дифузните интерстициални белодробни болести включват такива с позната етиология, каквито са колагено-васкуларните заболявния и свързаните с прием на медикаменти или вредности на околната среда, както и заболявания с непозната етиология, каквито са идиопатичните интерстициални пневмонии, грануломатозите и други белодробни заболявания с характерна хистологична картина (лимфангиолейомиоматоза, еозинофилна пневмония).
Идиопатичните интерстициални пневмонии (ИИП) са хетерогенна група болести, засягащи дифузно белодробния паренхим, при които се наблюдава увреждането му с наличие на вариращи модели на възпаление и фиброза. Интерстициумът, който включва пространството между епителните клетки и ендотелните базални мембрани, е първата локализация, която се уврежда при ИИП. При тези нарушения обаче често се засягат и периферните въздухоносни пътища, протежението на съдовете и техните епителни и ендотелни клетки1.
След като Liebow и Carrington описват за първи път идиопатичната белодробна фиброза (ИБФ), класификацията на тази група болести многократно се променя, като за описанието и дефинирането им в научната литература са налице много различни дефиниции и терминология, затрудняваща интердисциплинарната комуникация и сравняването на резултатите от провежданите клинични проучвния2, 3, 4.
Класификации на ИИП
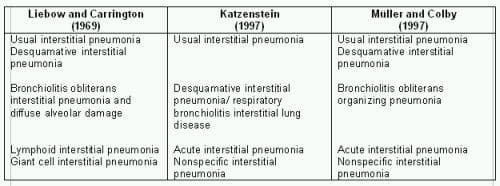
През 1969 г. Liebow и Carrington създават забележителната хистологична класификация на хроничните интерстициални пневмонии, като описват пет хистологични типа: обикновена интерстициална пневмония (UIP), интерстициална пневмония с облитериращ бронхиолит и дифузно алвеоларно увреждане, десквамативна интерстициална пневмония, лимфоцитна интерстициална пневмония и гигантскоклетъчна интерстициална пневмония2. На Табл. 1 са представени класификациите на ИИП, използвани до приемането на консенсусния документ на Американската торакална асоциация и Европейската респираторна асоциация (АТS/ERS)1.
През 2001 г. се провежда международна консенсусна среща на ATS и ERS, която има за цел да създаде единна класификация на интерстициалните белодробни фибрози, вземайки под внимание клиничните, рентгенологични и патологоанатомични аспекти на заболяването. Според този документ идиопатичните интерстициални пневмонии се класифицират като: идиопатична белодробна фиброза (IPF), неспецифична интерстициална пневмония (NSIP), криптогенна организираща се пневмония (COP), остра интерстициална пневмония (AIP), интерстициална белодробна болест, свързана с респираторен бронхиолит (RB-ILD), десквамативна интерстициална пневмония (DIP) и лимфоидна интерстициална пневмония (LIP)1. Без да бъде дефинирана самостоятелно, е призната и още една форма – некласифицираната интерстициална пневмония. Тази класификация заменя по-старите класификации на Liebow (1969)2, Katzenstein и Myers (1998)3. Новата класификация дефинира клиничното представяне, патологоанатомичните и рентгенологични белези при пациенти с ИБФ с основната цел да се стандартизира класификацията, да се уеднаквят дефинициите и критериите при поставянето на диагнозата идиопатична интерстициална белодробна болест. В тази класификация основно са представени хистологичните модели на клинико-рентгенологично-патологичната диагноза (Табл. 2).
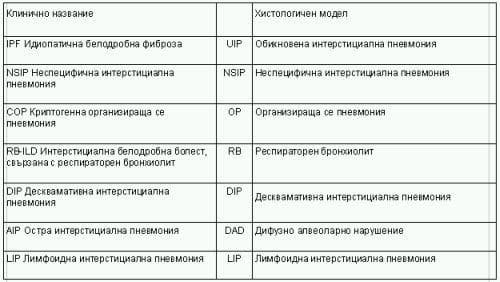
Основанието за избор на тези наименования и включването им във всяка форма е дискутирано подробно в консенсусния документ. С оглед изясняване на връзката между историческата, патологоанатомична и клиничната терминология, която се използва при описанието на тези форми, новата класификация дефинира понятието хистологичен модел, който поставя основата на окончателната клинико-рентгенологично-патологична диагноза (Табл. 2). Въпреки че хистологичните модели, оценявани от патолозите, разрешават по-доброто детайлизиране на формите на ИИП и осигуряват първичната база за разделянето на тези нарушения в различни категории, те служат основно за фундамент на класификацията. Все пак финалната клинико-патологична диагноза, включваща резултата и определяща типа нарушение – идиопатично или такова с позната етиология, може да се направи единствено след внимателно обсъждане на клиничните и рентгенологични промени. От това следва, че окончателната диагноза може да бъде оповестена, след като пулмолог, рентгенолог и патолог са обсъдили всички данни, свързани с пациента – клинични, рентгенологични и патологоанатомични.
Кое налага изработване на нова класификация на интерстициалните пневмонии
Различни разработки предизвикват и правят наложително изработването на нова изчерпателна клинико-рентгенологично-патологична класификация на ИИП по четири причини.
1. Публикуването на данни от големи групи пациенти с ИИП, съпроводени с патологоанатомична оценка от експерти в областта на белодробната патология, установява наличието на ясна картина на типа на хистопатологичния модел и неговата взаимовръзка с лечението4-7.
2. Наличието на по-малко инвазивнна хирургия под формата на видео-асистирана торакоскопска белодробна биопсия даде възможност на клиничните лекари да препоръчват на своите пациенти хирургичната биопсия8-10. Това доведе до повишаване броя на извършваните белодробни биопсии.
3. Широкото използване и подобреното разбиране на ползата от високоразделителната компютърна томография (HRCT) в оценката на тези заболявания доведе до подобряване на разбирането и представянето на разпространението и тежестта на лезиите9, 12-15.
4. Развитието на нови терапевтични подходи за лечението на фиброзните белодробни заболявания повиши интереса с оглед разбирането на патогенезата на тези нарушения16, 17.
В основата на класификацията на ATS/ERS стоят дефинирани морфологични модели, или т.нар. pattern, които са базирани на хистологични критерии и се асоциират с типични промени, които се демонстрират при осъществяването на HRCT. Въпреки тези дефинирани модели, на практика са налице препокриващи се форми, между отделните ИИП, което изисква приемане на принадлежност към един или друг подтип и тясна колаборация между клиницисти, рентгенолози и патолози. Тъй като обособените седем групи на ИИП имат различно клинично представяне, точната диагноза и диференциална диагноза са важна предпоставка за планиране на лечението и оценка на прогнозата.
Идипоатична белодробна фиброза (IPF, ИБФ) – usual interstitial pneumonia (UIP)
Това е най-честата форма на ИИП и е нарушението, което е свързано с най-лоша прогноза в дългосточен порядък (очаквана преживяемост след поставяне на диагнозата е средно 2.5-3.5 години). Според дефиницията клиничната картина се означава като ИБФ, докато морфологичният модел се обозначава като обикновена интерстициална пневмония (usual interstitial pneumonia, UIP). Моделът на UIP е характерен, но не е специфичен за ИБФ, като той може да бъде установен и при белодробна манифестация на колагенози, васкулити и увреждания на белия дроб, причинено от инхалация на различни вредни вещества. Мъжете боледуват по-често от жените, като по време на поставяне на диагнозата възрастта им обичайно е по-голяма от 50 години. Началото на болестта е почти незабележимо и се характеризира с поява на прогресиращ за няколко месеца задух1. По правило пациентите не показват клиничен отговор към приложението на високи дози кортикостероидни препарати (КС), но се наблюдава клиничен ефект при комбинирано приложение на КС и cyclosporine по време на фазата на обостряне.
Хистологичната картина на UIP се характеризира с изразена хетерогенност на белодробните промени. В един биопсичен материал в непосредствена близост могат да бъдат наблюдавани интактен белодробен паренхим и фибротични промени в различни стадии на развитие. Характерни за този модел са петнистите области от фиброза (т.нар. фибробластни огнища) и кистичните деструкции тип пчелна пита. При бързо прогресиране на заболяването могат да се открият промени, които са типични за дифузните алвеоларни нарушения (DAD) или за организиращата се пневмония. При пациенти с типично представяне на HRСТ-промени и клинични симптоми диагнозата UIP може да бъде поставена с вероятност 90-95%, като според някои автори дори не е необходимо допълнително хистологично изследване25. При разминаване в диагнозата и неадекватна клинична симптоматика или атипични HRСТ- промени е необходимо осъществяване на отворена белодробна биопсия. Определянето на подходящо място за биопсия става въз основа на HRСТ-промени, като се подбира периферно разположен участък с фиброза. Пациентите с ИБФ често имат значително повишен риск от белодробни карциноми, което изисква повишено внимание и СТ-контрол6. Подобни на UIP и често отграничими само по данни от клиничната информация хистологични картини могат да се наблюдават при азбестоза, колагено-васкуларни заболявания, екзогенен алергичен алвеолит, пострадиационна фиброза и при синдрома на Hermansky-Pudlak.
Неспецифична интерстициална пневмония
Поставянето на тази диагноза е особено диагностично предизвикателство, тъй като тя се представя с вариабилно клинично протичане и широк спектър от рентгенологични и хистологични промени. Поради тази хетерогенна манифестация на неспецифична интерстициална пневмония (НСИП) при описанието и дефинтрането й в научната литература няма единно становище и е твърде възможно в близко бъдеще тя да бъде подложена на нова класификация. Поради това се препоръчва тя да се възприема като „провизорна” диагноза до натрупването на нови знания1. Пациентите с НСИП са обичайно на възраст между 40 и 50 години или с около декада по-млади от тези с ИБФ. Липсва различие на заболеваемостта между мъже и жени, както и връзка между развитието на болестта и тютюнопушене. Сравнени е пациентите с ИБФ, тези с НСИП имат по-добра прогноза и в по-голямата си част показват подобрение или поне стабилизиране на симптоматиката под въздействието на КС терапия18. В зависимост от хистологичния подтип преживяемостта в най-добрия случай не се повлиява, а в най-лошия случай е между 6 и 13 години19.
Хистологичния модел на NSIP се характеризира с хомогенност на белодробните поражения във времето и пространството. Това я отличава значително от хетерогенната хистологична картина на UIP. В зависимост от това дали в хистологичната картина доминират възпалителни клетки, или зони с фиброза, НСИП се определя като целуларен или фибротичен подтип. Целуларната форма се наблюдава по-рядко от фибротичната, но има значително по-добра прогноза и по-добър отговор към лечение с КС20.
Поради припокриващите се HRСТ-морфологични промени UIP е важна диференциална диагноза на НСИП и при съмнение за нея е задължително хистологично потвърждение. Различни проучвания показват, че от всички ИИП диагнозата НСИП се поставя най-трудно както от патолози, така и от рентгенолози21, 22, като това е валидно в по-голяма степен за фибротичния подтип, който трудно се разграничава от UIP, и често окончателната диагноза се поставя в контекста на клиничното протичане. Хитологичната картина на НСИП може да бъде установена при наличие на умерено интерстициално хронично възпаление, рехава гранулационна тъканна или плътна интерстициална фиброза, съставена от колагенни влакна, или комбинация от тези промени. Характерна особеност е подчертано петнистото разпределение, което с времето преминава в хомогенна съединителна тъкан (рехава или плътна) с еднаква давност4, 5, 24. Дори и в зоните с налична фиброза белодробната архитектоника е запазена за разлика от UIP. Ако в основата на увреждането стои възпаление, е необходимо да се направи хистологична диференциална диагноза с оглед разграничаването на НСИП от екзогенен алергичен алвеолит, колагено-васкуларно заболяване, медикаментозно индуцирани белодробни заболявания (напр. еозинофилна пневмония), от инфекция или имунен дефицит. При фиброза диференциалната диагноза изключва наличието на стар екзогенен алергичен алвеолит (активирани зони), саркоидоза (грануломи) или грануломатоза от Лангерхансови клетки (активни огнища).
Криптогенна организираща се пневмония
Тя е добре характеризирана форма с типична клинична, рентгенологична и хистологична картина. По-рано беше обозначавана като идиопатична бронхиолитис-облитеранс организираща се пневмония (ВООР), но това понятие бе изоставено, за да се избегне объркване с други заболявания на дихателните пътища, като констриктивен бронхиолит. Морфологичният модел на криптогенна организираща се пневмония (КОП) е организиращата се пневмония (ОР), която в своята идиопатична форма (в рамките на КОП) се среща рядко. По-често се среща като вторично нарушение, свързано с колагенози, инфекциозно и медикаментозно индуцирани белодробни заболявания25. Не съществува разлика между засягането на мъже и жени, най-честото проявление е между 50 и 60 години. Непушачите заболяват два пъти по-често от пушачи. Началото на заболяването е подостро, за по-малко от три месеца. Пациентите с КОП показват отличен отговор към лечение с КС, което по правило продължава около три месеца. След преустановяване на лечението не рядко се достига до рецидиви, които отново трябва да бъдат лекувани с КС22.
Хистологично ОР се характеризира с гранулационно-тъканни образувания, които изпълват полипообразно алвеоларните ходове и алвеолите, без да водят до нарушение на белодробната архитектура. Друга характеристика на ОР е тенденцията за миграция, при което паренхимните алтерации могат да променят не само местоположението си, но и морфологията си. Заедно с тези типични СТ-манифестации на ОР са познати и атипични модели, като множествени възли, нерегулярни линеарни задебеления, които при солитарна манифестация могат да имитират бронхогенен карцином25. Независимо от морфологията, при всички случаи на КОП е индицирано хистологичо изследване. Още не е достатъчно добре оценено дали осъществяването на трансбронхиална биопсия е достатъчно за диагнозата.
Въз основа на много честата картина на реакция на образуване на гранулационна тъкан в белия дроб при този модел диференциалната диагноза е много широка. Срещу наличието на КОП свидетелства наличието на неутрофилни гранулоцити в алвеолите, налчието на остър бронхиолит, грануломи, некрози, хиалинни мембрани, изобилие от фибрин в алвеоларните пространства, васкулит и особено увеличен брой еозинофилни гранулоцити (еозинофилна пневмония)23. С изключение на лесно разграничимата UIP, по-често се налага при хистологичната диференциална диагноза да се обсъжда модела на DAD, по-рядко модела на NSIP или DIP. Преди да бъде поставена клиничната диагноза КОП трябва да бъдат изключени редица заболявания, протичащи с хистологичен модел на ОР (организиращи се инфекции, колагено-васкуларни заболявания,увреждания, причинени от инхалация на вредни вещества, медикаментозно индуцирани реакции и др.).
Остра интерстициална пневмония
Тя се различава клинично от останалите форми на ИИП по своето остро (в рамките на няколко дни) и бързо прогредиентно симптомно клинично протичане. В предходни класификации острата интерстициална пневмония (ОИП) бе наричана синдром на Hamman-Rich. Клинично, рентгенологично и хистологично промените при ОИП отговарят на тези при остър респираторен дистрес синдром (acute respiratory distress syndrome, ARDS). Понятието ОИП е резервирано за идиопатични случаи, докато ARDS би трябвало да се използва при случаи с клинично позната етиологична причина. Пациентите с ОИП показват широко възрастово разпределение. Често острото протичане поставя на преден план въпроса за наличие на инфекция на дихателните пътища. По правило бързо се достига до дихателна недостатъчност и въпреки механичната вентилация и другите интензивни подходи при лечение леталитетът надвишава 50%1. Позитивен ефект от лечение с КС се наблюдава изключително в ранните фази на болестта.
Хистологичният модел на ОИП отговаря на дифузно алвеоларно нарушение (diffuse alveolar damage, DAD) и поради това не се отличава от хистологичната картина при ARDS. В острата или ексудативна фаза се достига до образуване на алвеоларен оток и хиалинни мембрани, във фазата на организация се установява хиперплазия на пневмоцити и рахаво фибротично разширение на алвеоларните септи. Хистологичният DAD-модел показва предимно дифузни, с еднаква давност промени, характеризиращи се с оток, хиалинни мембрани и остра възпалителна интерстициална инфилтрация в ексудативна фаза. Пролиферативната фаза се характеризира с наличие на рехава гранулационна тъкан, предимно в интерстициума, мозаични оскъдни остатъци от хиалинни мембрани и пролиферирали пневмоцити-тип ІІ. При по-нататъшното протичане могат да се открият участъци на нормален белодробен паренхим или фиброза, която може да достигне до фиброкистично преустройство29, 30. При HRCT-изследвне в ранните фази се установявт двустранни промени тип матово стъкло, при което разположението на лобулите показва характерен географски модел28. В ангажираните участъци се откриват типични области на консолидация. В стадия на организация се откриват тракционни бронхиектазии и разрушаване на белодробната архитектура. При малкото пациенти, преживели ОИП, се наблюдава прогредиентно обратно развитие на промените тип матово стъкло и областите на консолидация. Остатъчните промени включват ретикуларни структури, малки кисти и увредена белодробна архитектура. Образната морфология на HRСТ се покрива с ARDS, но при ОИП се наблюдава симетрично засягане на долните белодробни дялове и във фибротичния стадий се установява наличие на промени тип пчелна пита.
Хистологичната диференциална диагноза се базира на липса на грануломи, некрози, вирусни включвания, различни микробни причинители, микроабсцеси, които са белег на инфекция. Наред с това трябва да липсват промени, характерни за UIP в стадия на бързата прогресия, с наличие на еозинофилни гранулоцити (еозинофилна пневмония), екзогенен алергичен алвеолит или васкулит (колагено-васкуларно заболяване). Други важни диференциално-диагностични тези обхващат медикаментозно-токсични увреждания, уремия, сепсис, трансфузионни нарушения, шок и травма.
Десквамативна интерстициална пневмония
Интерстициална белодробна болест, свързана с респираторен бронхиолит (ИББ-РБ), и десквамативна интерстициална пневмония (ДИП). Това са две форми на ИИП, които се срещат почти изключително у пушачи. Първата е клиничен вариант на често и случайно диагностицирания хистологично респираторен бронхиолит при пушачи. Втората протича клинично с утежняване на симптомите и се разглежда като краен стадий на ИББ-РБ. Мъжете боледуват два пъти по-често от жените, като заболяването се проявява между 40- и 50-годишна възраст. Пациентите обичайно са дългогодишни пушачи. Клиничната картина се характеризира с бавно прогресиращ задух и дразнеща кашлица. След преустановяване на тютюнопушенето се наблюдава частична обратимост на промените и добра прогноза. В редки случаи при ДИП се достига до дихателна недостатъност. Хистологично и при двете форми се намират голямо количество пигментирани алвеоларни макрофаги, като при ИББ-РБ те са разположени бронхиолоцентрично, а при ДИП – интраалвеоларно. Паралелно с това се демонстрира ограничена интерстициална фиброза. Определението десквамативна интерстициална пневмония е въведено въз основа на старите представи за наличие на т.нар. десквамирани пневмоцити, които обаче могат да се намерят много рядко. При HRCT картината на ИББ-РБ се демонстрира с дифузно нарушение или доминиращо ангажиране на горните белодробни дялове, докато ДИП ангажира доминиращо долните дялове и субплевралните области. Други HRCT-белези за ИББ-РБ са центрилобуларните нодули (airspace nodules), които представляват малки възелчета с плътност на матово стъкло31, 32. Белег са и дифузните промени тип матово стъкло, задебеляването на бронхиалната стена и съпровождащият ги центроацинарен белодробен енфизем. От своя страна, ДИП се характеризира с дифузни и/или периферни промени тип матово стъкло. В тези зони могат да се образуват малки кисти, като за разлика от кистичните промени при UIP, тук белодробната архитектура остава непроменена4. Въз основа на клиничното и рентгенологичното припокриване между ИББ-РБ и ДИП за прецизиране на диагнозата се препоръчва отворена белодробна биопсия.
Хистологичната диференциална диагноза на RB включва DIP, бронхиолит и НСИП. Моделът на RB и DIP-моделът представят в крайна сметка един спектър от нарушения и често могат да се припокриват28. Хистологичната диференциална диагноза на DIP включва локални неспецифични DIP-подобни реакции около други огнищни процеси (тумори, Лангерханс-клетъчна грануломатоза и др.). Срещу UIP говори липсата на фиброзно-кистична преустройка и фибробластни огнища.
Лимфоидна интерстициална пневмония
Включването й към групата на ИИП се дискутира широко, тъй като идиопатичната й форма се среща рядко. По-често моделът на лимфоидна интерстициална пневмония (ЛИП) се среща като вторично нарушение, свързано с автоимунни болести и особено със синдрома на Sjоgren и с имуносупресивни заболявания, като HIV инфекция. Засяга по-често жени на възраст около 50 години. Клиничните симптоми по правило се характеризират с лек задух и кашлица. При вторични форми на ЛИП на преден план излизат симптоми на основното заболяване, като белодробните промени са най-често случайна находка. При ЛИП прогнозата най-често е добра и само в единични случаи се наблюдава прогресия до белодробна фиброза. В много редки случаи може да премине в малигнен лимфом33. Терапевтичният подход включва приложението на КС, въпреки че ефективността от приложението им все още е спорно.
Хистологичният модел на ЛИП е дифузна инфилтрация на алвеоларните септи от лимфоцити, плазмоцити и макрофаги. Допълнителна находка са лимфните фоликули в перибронхиалните области. При HRCT се представят промени тип матово стъкло, които или засягат дифузно двете белодробни половини, или ангажират белодробните основи34. Характерен, но не константен белег за ЛИП са периваскуларните тънкостенни кисти, които могат да доведат до деструкция на бронхиолите35. Могат да се наблюдават и ретикуларни промени, центрилобуларни нодули и задебелени интерлобуларни септи.
Хистологичната диференциална диагноза включва лимфоми (имунохистохимия и молекулярна биология) и екзогенен алергичен алвеолит (ограничено и по-изразено бронхиолоцентрично възпаление, грануломи, гранулационна тъкан), както и хистологичен модел на ОР, NSIP и UIP23. При позната етиологична причина тази картина може да се наблюдава при инфекции, предизвикани от Pneumocystis carinii, Hepatitis B, Epstein-Barr вирус и др., колагено-васкуларни заболявания (синдром на Sjоgren, ревматоиден артрит, системен еритематоден лупус), имунни дефицити и други имунологични болести, както и медикаментозно-токсично индуцирани нарушения.
Значение на клинично-рентгенологично-патологоанатомичната находка при поставяне на диагноза
Класификацията на ATS и ERS съдържа седем различни форми с дефинирани морфологични модели, които се базират върху хистологични критерии и се асоциират с промени при HRCT-изследване. С изключение на ясния при клинично и HRCT-изследване модел на UIP, при който диагнозата се поставя лесно, всички останали форми изискват детайлно хистологично изясняване. При всички форми се налага извършването на отворена белодробна биопсия. Предпочитат се биопсии от повече от един белодробен дял36, 37, 38. С оглед оптимална оценка е необходимо изпращането на биопсичния материал (стерилен и нефиксиран) до патологоанатомичната лаборатория да става по възможно най-бърз начин. Като алтернатива е възможно след вземане на биопсия върху материала да бъде аплициран разтвор за фиксация, като във всички случаи трябва да се предотврати развитие на ателектаза. Трансбронхиалната биопсия по правило служи за изключване на саркоидоза, неоплазми и определени инфекции. Понякога, за да се постави диагноза, е достатъчно наред с типичната клинико-рентгенологична картина за КОП или ОИП да се установи с трансбронхиална биопсия хистологичен модел на OP или DAD. Ако това не е достатъчно за поставяне на диагнозата, на по-късен етап се обсъжда осъществяване на отворена хирургична биопсия. Бронхоалвеоларният лаваж намира място като подпомагащ диагнозата метод, в зависимост от клинико-рентгенологичния образ и диагностицираното заболяване, както и при редица дифузни интерстициални белодробни болести (саркоидоза, екзогенен алергичен алвеолит, еозинофилна пневмония, ИББ-РБ, ДИП и др.). Въз основа на различната прогноза и различното лечение на интерстициалните белодробни болести преди началото на третирането пациентите трябва да бъдат диагностицирани и поставени в една от посочените по-горе подгрупи. Този подход обаче е възможен единствено при тясно сътрудничество и динамична, свързана с диагностичния процес, екипна работа между пулмолози, рентгенолози и патолози, насочена както в полза на пациента, така и с оглед оптимизиране на консенсусната класификация на интерстициалните белодробни болести.
д-р Диана Петкова, дм, гл. асистент, Клиника по пневмология и фтизиатрия, УМБАЛ “Св. Марина” - Варна, МУ “Проф.д-р Параскев Стоянов” – Варна
Литература
American Thoracic Society/European Respiratory Society. International multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304
2. Liebow AA, Carrington CB. The interstitial pneumonias. In: Simon M, Potchen EJ, LeMay M, editors. Frontiers of pulmonary radiology, 1st edition. New York: Grune&Stratton,1969; p. 102–141
3. Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998; 157: 1301–1315
4. Müller NL, Colby TV. Idiopathic interstitial pneumonias: high-resolution CT and histologic findings. Radiographics 1997; 17: 1016–1022
5. Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998; 157: 199–203
6. Nagai S, Kitaichi M, Itoh H, Nishimura K, Izumi T, Colby TV. Idiopathic nonspecific interstitial pneumonia/fibrosis: comparison with idiopathic pulmonary fibrosis and BOOP. Eur Respir J 1998; 12: 1010–1019
7. Schwartz DA, Van Fossen DS, Davis CS, Helmers RA, Dayton CS, Burmeister LF, Hunninghake GW. Determinants of progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1994; 149: 444–449.
8. Travis WD, Matsui K, Moss JE, Ferrans VJ. Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns. Survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol 2000; 24: 19–33
9. Nasim A, Akhtar RP, Spyt TJ. Video-thoracoscopic lung biopsy in diagnosis of interstitial lung disease. J R Coll Surg Edinb 1995; 40: 22–24
10. Lewis RJ, Caccavale RJ, Sisler GE, Mackenzie JW. One hundred consecutive patients undergoing video-assisted thoracic operations. Ann Thorac Surg 1992; 54: 421–426
11. Nishimura K, Kitaichi M, Izumi T, Nagai S, Kanaoka M, Itoh H. Usual interstitial pneumonia: histologic correlation with high-resolution CT. Radiology 1992; 182: 337–342
12. Shah SS, Tsang V, Goldstraw P. Open lung biopsy: a safe, reliable and accurate method for diagnosis in diffuse lung disease. Respiration 1992; 59: 243–246
13. Orens JB, Kazerooni EA, Martinez FJ, Curtis JL, Gross BH, Flint A, Lynch JP III. The sensitivity of high-resolution CT in detecting idiopathic pulmonary fibrosis proved by open lung biopsy. A prospective study. Chest 1995; 108: 109–115
14. Holt RM, Schmidt RA, Godwin JD, Raghu G. High resolution CT in respiratory bronchiolitis-associated interstitial lung disease. J Comput Assist Tomogr 1993; 17: 46–50
15. Swensen SJ, Aughenbaugh GL, Myers JL. Diffuse lung disease: diagnostic accuracy of CT in patients undergoing surgical biopsy of the lung. Radiology 1997; 205: 229–234
16. Hunninghake GW, Zimmerman MB, Schwartz DA, King TE, Lynch J, Hegele R, Waldron J, Colby T, Muller N, Lynch D, et al. Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2001; 164: 193–196
17. Ziesche R, Hofbauer E, Wittmann K, Petkov V, Block LH. A preliminary study of long-term treatment with interferon gamma-1b and low-dose prednisolone in patients with idiopathic pulmonary fibrosis. N Engl J Med 1999; 341: 1264–1269
Коментари към Клиничен обзор върху идиопатичните интерстициални пневмонии