Клинична класификация на пулмоналната хипертония
През 1998 г. на Втория световен симпозиум по пулмонална хипертония (ПХ) в Евиан, Франция, е предложена клинична класификация. Нейна цел е да индивидуализира различните категории, разделяйки сходствата в патофизиологичните механизми, клиничното представяне и терапевтичните възможности. Класификацията от Евиан е общоприета и широко разпространена в клиничната практика, особено в специализираните центрове. Използва се от Американската агенция по храни и лекарства и Европейската агенция за лекарствена оценка за етикетиране на новоодобрени медикаменти за ПХ (Табл. 1).
таблица 1. Клинична класификация на ПХ, Евиан, 1998.


През 2003 г. на Третия световен симпозиум по ПХ във Венеция, Италия, е взето решение да се поддържа общата структура и философия на класификацията от Евиан (Табл. 2).
Предложени са някои модификации:
(1) замяна на термина първична пулмонална хипертония с идиопатична пулмонална хипертония;
(2) рекласификация на белодробната вено-оклузивна болест и белодробната капилярна хемангиомиоматоза;
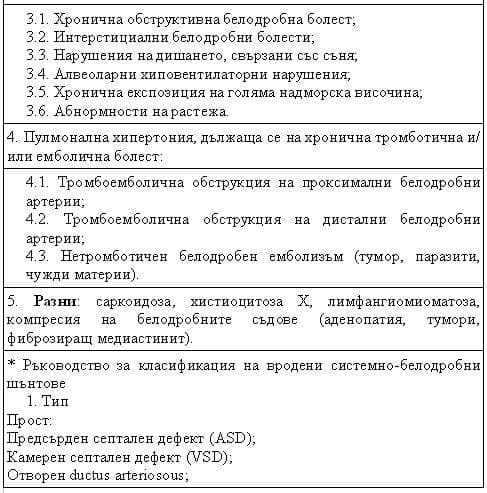
(3) обновяване на рисковите фактори и свързаните с ПХ състояния и предложение за ръководство за подобряване на класификацията на вродените системно-белодробни шънтове.
Таблица 2. Ревизирана клинична класификация на ПХ, Венеция, 2003.


Класификацията от Евиан съдържа пет категории, в които болестите с ПХ са групирани според специфични терапевтични интервенции, отправени към причината:
(1) пулмонална артериална хипертония (ПАХ);
(2) пулмонална венозва хипертония;
(3) ПХ, свързана с нарушения на дихателната система или хипоксемия;
(4) ПХ, причинена от тромботични или емболични болести и
(5) ПХ, причинена от болести, засягащи белодробните съдове. Всяка категория съдържа подгрупи, разделени в зависимост от причините и местата на увреждане.
Пулмонална артериална хипертония
Първата категория, наречена ПАХ, включва първата подгрупа без определени причини или т.нар. ППАХ (първична пулмонална артериална хипертония). Тук влизат фамилни и спорадични форми на болестта. Втората подгрупа включва няколко състояния или болести с познати причини, които започват от лезии в малките белодробни мускулни артериоли. Сред тях са лекарствено свързана ПХ, порто-пулмонална хипертония, HIV-свързана ПХ, колагенни съдови болести, вродени системно-пулмонални шънтове и персистираща ПХ при новороденото.
Въпреки че механизмите, отговорни за ремоделиране на белодробните артериоли при тези състояния са непознати, те споделят сходни морфологични особености, клинично представяне и клиничен отговор към лечение с продължителна инфузия на epoprostenol (особено ППАХ и ПАХ, свързана със склеродермия).
Пулмонална венозна хипертония (ПВХ)
Тази категория съдържа предоминантно левостранни клапни или миокардни болести, изискващи лечение, насочено към подобрение на миокардната функция или облекчаващо клапните механични дефекти по-бързо от белодробната вазодилататорна терапия. Възможно е лечението с epoprostenol при пациенти с ПВХ дори да навреди. Тази категория включва също външна компресия на белодробните вени и белодробна венооклузивна болест, която клинично имитира ППАХ.
Пулмонална хипертония, свързана с нарушения на дихателната система или хипоксемия
В тази категория доминираща причина е неадекватната оксигенация на артериалната кръв, като резултат от белодробни болести, нарушен контрол на дишането или пребиваване на голяма надморска височина. Тук нарастването на средното пулмонално артериално налягане (СПАН) обикновено е умерено (< 35 mm Hg). По правило преживяемостта зависи по-скоро от тежестта на белодробната болест, отколкото от белодробната хемодинамика. Продължителната кислородна терапия (16 или 24 h/денонощие) подобрява преживяемостта при пациенти с ХОББ. При хора, родени и развили ПХ на голяма надморска височина, преместването на морското равнище бързо подобрява ПХ и свързаните с нея симптоми.
Пулмонална хипертония, причинена от тромботична или емболична болест
Тази категория включва хронична тромбоемболична ПХ, дължаща се на проксимален организиран тромб в големи белодробни артерии, който може да се подобри от пулмонална ендартектомия, или повече периферни емболи или тромби, които са неразличими от тромботичните лезии, наблюдаване при ППАХ и могат да се лекуват с хронична белодробна вазодилататорна терапия. При всички случаи е препоръчителна продължителна антикоагулантна терапия.
Пулмонална хипертония, причинена от болести, засягащи белодробните съдове. Тази категория включва ПХ, произхождаща от възпалителни процеси или механична обструкция (напр. шистозомиаза, саркоидоза). Пулмоналната капилярна хемангиоматоза също е включена в тази група, въпреки че обичайно се представя клинично като венооклузивна болест.
Генетична класификация на ПХ
В светлината на настоящия напредък в разбирането на генетичната основа на ППАХ се обмисля нейна генетична класификация. Мутациите в гена, кодиращ рецептора за костния морфогенен протеин тип II (BMPR2), локализиран в хромозома 2q33, вероятно са в основата на приблизително 50% от случаите с ППАХ. Въпреки че остоналите 50% от семействата показват някои доказателства за връзка с локуса на BMPR2, не са идентифицирани специфични мутации в кодовия или промотиращия регион. Освен това, мутациите в BMPR2 са открити в при повече от 26% от спорадичните случаи на ППАХ. Въпреки че някои от тези случаи могат да възникнат отново от мутации, повечето представляват фамилна трансмисия на мутантния BMPR2 с ниска пенетрантност на гена за болестта. Същевременно има доказателства за втори локус в 2q31, въпреки че той е от типа mapped и обуславя по-скоро фенотип с абнормен белодробен съдов отговор към физически усилия, отколкото да се манифестира при ППАХ. Мутациите в ген BMPR2 изглеждат напълно специфични за т.нар. ППАХ, но същевременно са идентифицирани в редки случаи на ПАХ, свързана с прием на апетито-потискащи лекарства и венооклузивна болест. Досега търсенето на BMPR2 мутациите при други форми на ПХ е негативно.
Генетичните проучвания показват, че мутациите в BMPR2 не са достатъчни сами по себе си, за да предизвикат клинично изявена болест. От това следва, че шансът за генно обусловено клинично развитие на ППАХ е по-нисък от 20%. Това наблюдение подчертава критичната роля на други генетични/външни фактори в обсъждането на чувствителността към ПХ.
Необходими са още проучвания за идентифициране на други гени, модификатори и регулаторни гени за ПХ и да се определи дали пациентите с ПАХ и мутации на BMPR2 са различни от пациенти с ПАХ без мутации по отношение на отговора към лечение, възрастта на възникване, тежестта и естествения ход на болестта.
Рискови фактори и състояния
Рисков фактор за ПАХ е всеки признак или състояние, което се подозира в предразполагащ или улесняващ аспект. Рисковите фактори могат да включват лекарства и химикали, болести или фенотип (възраст, пол). Терминът асоциирани състояния се използва, когато не е възможно да се определи дали предразполагащият фактор е налице преди началото на ПХ. Тъй като наличието на „абсолютен” риск от известните рискови фактори за ПАХ е ниско, вероятно играят важна роля индивидуалната чувствителност или генетичната предиспозиция.
По време на срещата в Евиан (Табл. 3) различните рискови фактори и асоциирани състояния са категоризирани според връзката им с ПАХ и тяхната вероятна причинна роля:
(1) като определени се дефинират състояния, включени чрез наблюдения на големи контролирани проучвания или недвусмислени епидемии;
(2) много вероятни са тези, определени в някои наблюдения, включително и в широки серии от случаи и проучвания;
(3) като възможни се означават такива, свързани и основани на серии от случаи, регистри или експертни мнения; (iv) като невероятни се означават подозрителни рискови фактори, за които контролираните проучвания не демонстрират някаква корелация.
Таблица 3. Рискови фактори и асоциирани състояния за ПАХ, класифицирани според силата на доказателствата, Евиан, 1998.


Милена Енчева, д-р, ст. асистент, Клиника по белодробни болести, Военномедицинска академия – София
Литература
Simonneau G, Nazzareno G, Rubin L, et al, Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43: 5S-12S
Fishman AP. Clinical classification of pulmonary hypertension. Clin Chest Med 2001; 22: 385-91
Humbert M, Nunes H, Sitbon O, et al. Risk factors for pulmonary arterial hypertension. Clin Chest Med 2001; 22: 459-75
Olshewski H, Simonneau G, Galie N, et al. Inhaled iloprost in severe pulmonary hypertension. N Engl J Med 2002; 347:322-7
Коментари към Клинична класификация на пулмоналната хипертония