Идиопатична пулмонална артериална хипертония
Идиопатичната пулмонална артериална хипертония (ИПАХ), наричана до скоро първична белодробна хипертония (ПБХ), е рядка болест, хартеризираща се с повишено белодробно артериално налягане, за което липсва видима причина. Тя се обозначава още като прекапилярна пулмонална хипертония. Диагнозата се поставя след изключване на други познати причини, водещи до пулмонална хипертония (ПХ). Dresdale et al. за първи път описват хемодинамичните промени при ИПАХ през 1951 г. В норма средното пулмоартериално налягане (СПАН) се движи между 12-16 mm Hg. Според регистъра на Националният институт на здравето в Америка пулмоналната артериална хипертония (ПАХ) се дефинира като повишено налягане в белодробната артерия (БА) над 25 mmHg при покой и над 30 mm Hg след натоварване, след изключване на други познати причини, включващи левостранно клапно сърдечно заболяване, инфаркт на миокарда, вродено сърдечно заболяване, септален дефект, заболяване на съединителната тъкан, хронично тромбоемболично заболяване, тежко хронично обструктивно белодробно заболяване1.
Клинично представяне на ПХ
В миналото ПАХ обичайно се каласифицираше като първична (идиопатична ПХ) и постпървична (вторична). Терминът първична ПХ беше съхранен в класификацията от Евиан, Франция, поради широката му употреба. През 2003 г. по време на Третият световен симпозиум по ПАХ, проведен във Венеция, Италия, бе въведена третата преработена класифиакация на СЗО, разделяща ПАХ в пет основни категории. На този форум бе оценена класификацията на ПАХ от Евиан и бяха предложени някои модификации, приета бе т.нар. Венецианска клинична класификация на пулмоналната хипертония с оглед по-добро разбиране и по-лесна употреба. С цел да се избегнат възможни обърквания във Венеция бе решено, че първата категория, означена като пулмонална артериална хипертония (ПАХ) трябва да включва три основни подгрупи: (1) идиопатична пулмонална артериална хипертония (ИПАХ), (2) фамилна пулмонална артериална хипертония (ФПАХ) и (3) пулмонална артериална хипертония, свързана с рискови фактори (РФ) или асоциирани състояния (АПАХ).
При редица болести, като портална хипертония2-4, инфекция с HIV5-7 и прием на апетитопотискащи медикаменти, като aminorex, fumarat, fenfluramine8-10 се установяват патологични промени, подобни на тези при ИПХ. Публикувани са съобщения, които коментират връзката на ИПАХ по време на бременност или при прием на орални контрацептивни медикаменти11-13. През 1981 г. при консумация на репично масло в Испания се установява повишаването на честотата на ИПАХ при 20% от всички пациенти14. Hoeper et al. съобщават за наличие на ИПАХ в периода от 4 до 34 години след осъществяване на спленектомия15.
Честота на ИПАХ
Среща се рядко, честотата й се изчислява на 2 случая за 1 млн. население за година. В Америка ИПАХ е отговорна за смъртта на 125-150 души годишно, като заболеваемостта се изчислява на 1-3 случая годишно за 1 милион население. Заболеваемостта и смъртността от ПАХ са значително по-високо от тези при чиста ИПАХ. Световната заболеваемост от ИПАХ се равнява на тази, изчислена за Америка. Абсолютният риск при лица със затлъстяване, които са приемали апетитопотискащи медикаменти за повече от три месеца, е над тридесет пъти по-голяма в сравнение с тази при общата популация16,17.
Расови особености.
Честотата на ИПАХ не показва расови различия, но при чернокожата раса се наблюдава по-високо съотношение между жени и мъже – съответно 4.3:1.
Пол. ИПАХ се среща по-често при жени, отколкото при мъже, като съотношението жени към мъже се отнася, както 1.7:1 до 3.06:118. В зависимост от данните на различните центрове съотнощението жени към мъже се движи от 2:1 до 9:1. Остава неясно защо женският пол се явява предразполагащ фактор.
Възраст. По правило ИПАХ се проявява при млади жени в детеродна възраст, обаче може да се регистрира във всяка възраст, дори и в детска. С най-голяма честота се появява между третата и четвъртата декада от живота, като средната възраст на поставяне на диагнозата според редица студии е 36 години1,19, 20. Наблюдава се тенденция към по-голяма възраст на поставяне на диагнозата при мъже, сравнено с жени. Наблюдават се е и случаи при жени на възраст 50, 60 или повече години.
Наследственост
Позната е фамилна форма на ИПАХ, която се изчислява на 6% от всички случаи. Досега са описани два генетични дефекта, свързани с ПАХ. Мутацията в гена за рецептора на костния морфогенен протеин тип II (BMPR2), разположен върху хромозома 2q33, води до развитие на ПАХ при около 20% от засегнатите8. Унаследяването е автозомно доминантно с непълна пенетрация на гена. Установени са и спорадични форми. Дефекти в ALK-1-гена, заедно с хередитарната форма на телеангиектазии (болест на Osler-Rendu-Weber), могат да доведат и до развитието на ПАХ, която се доближава клинично до ИПАХ и фамилна ПХ.
Много съвременни изследвания са насочени за установяване на молекулярните механизми на ИПАХ. Идентифицирани са мутации в региона на хромозома 2q33, при които се позитивира гена на костния морфогенен протеинов рецептор тип II (BMPR2)24. Костните морфогенни протеини са част от фамилията на трансформиращите растежни фактори (TGF-β) – циркулиращи протеини, които регулират растежа и възстановяването на тъканите във всички органи. Нормалната функция на BMPR2 в белодробната циркулация вероятно е антипролиферативна25, 26. Вероятно при много случаи на ИПАХ, както при спорадична, така и при фамилна форма, болестта се причинява първично от дефект в BMPR2, но при хетерозиготи мутацията само на BMPR2 е недостатъчна, за да предизвика клинично проявена болест. Необходим е т.нар. вторичен соматичен генетичен удар и той вече е мутация в TGF-β-рецептора, която може да доведе до развитие на ИПАХ. Клиничните и патологоанатомични промени, както и преживяемостта след поява на симптомите, не се различават от тези при нефамилната ИПАХ. В света са познати 94 семейства, при които е установено автозомно-доминантното унаследяване с различна пенетрантност на гена. Рискът на едно лице, принадлежащо към тези фамилии, да развие ИПАХ е 0.6-1.2%.
Aсоциирани състояния
ИПАХ може да бъде асоциирана с портална хипертензия (често наричана портопулмонална хипертония), което води до заключението, че пациенти с наличие на шънтиране на кръв от спланхниковата система (със или без наличие на чернодробно заболявне) са с по-висок риск за развитието на ИПАХ. В допълнение, излагането на белодробната циркулация към субстанции от спланхниковата циркулация, която нормално претърпява дезинтоксикация в черния дроб, може да доведе до развитие на ПХ. В тази посока са небходими допълнителни изследвания за по-добро разбиране на посочените механизми. Приема се, че наличието на съединителнотъканни заболявания, като CREST синдром (калциноза на кожата, Raynaud феномен, нарушения в мотилитета на хранопровода, склеродактилия, и телеангиектазия) например, варианти на склеродермия, системен еритематоден лупус и смесена съединителнотъканна болест, е предразполагащ фактор за развитието на патологично състояние, подобно на ИПАХ. В новата класификация на ПАХ тези състояния са дефинирани като асоциирани с ПАХ болести.
Патофизиологичната основа на предразполагащите състояния е неясна. Други събития, свързани с ПАХ, включват експозиция на анорексигени и други алфа-адренергични стимуланти (например кокаин, амфетамини), както и позитивна серология за НIV. По какъв начин те водят до развитие на ПХ все още не е достатъчно добре изяснено. В повечето случаи честотата на асоциираната ПХ, например, при наличие на колагеносъдови болести е по-висока (> 10%) в сравнение с ИПАХ, чиято заболеваемост се изчислява на 1-2 случая за милион население годишно.
Ход на болестта
Най-общо пациентите с ИПАХ имат лоша прогноза. В Холандия средната продължителност на живот след първа визита при общопрактикуващ лекар е 4.6 години, докато средната продължителност на предстоящия живот към момента на поставяне на диагнозата е 2.8 години. При някои от случаите се установява по-голяма преживяемост поради спонтанна регресия на болестта. Пациенти, които отговарят с вазодилатация по време на тест с калциев антагонист и в последствие се лекуват с медикаменти от тази група, имат по-висока преживяемост, отколкото пациенти, третирани дългосрочно с интравенозен epoprostenol. Качеството на живот при тях се запазва или се подобрява.
Болестност/смъртност
ИПАХ е заболяване, за което няма лечение. Без медикаментозно лечение ИПАХ води до десностранна сърдечна недостатъчност и смърт. В едно проучване общата преживяемост на проследените пациенти се изчислява на 30% за период от три години. Проучвания, докладващи резултати от аутопсия на пациенти, показват болестност в 0.13%. При пациенти с чернодробна цироза болестността е 0.73%2.
За периода до 1990 г. наличните терапевтични възможности бяха ограничени. След появата на простациклиновите аналози, ендотелин-рецепторните антагонисти, фосфодиестеразните инхибитори и други нови медикаменти прогнозата на пациенти с ИПАХ и заболявания, асоциирани с ИПАХ, се подобри значително. Проучвания, обобщаващи резултати от дълготрайно приложение на тези групи медикаменти, като монотерапия или в схеми за комбинирано приложение, показват повишаване на петгодишната преживяемост до 65%. Очаква се, че комбинираното приложение на новите медикаменти ще доведе до още по-добри бъдещи резултати.
Хистопатология
Различни хистологични модели се свързват с белодробна артериопатия при ИПАХ, като една от тях включва тромбози in situ. Тромботичната белодробна артериопатия може да бъде наблюдавана със или без наличие на плексиформени лезии. Тя се характеризира с тромбози in situ на малките мускуларни артерии от белодробното съдово легло. Тромботичната белодробна артериопатия често се представя като най-ранен стадий на ИПАХ (т.е. преди развитие на плексогенна белодробна артериопатия) или като необратими лезии в по-късен стадий. Наблюдава се повишаване на циркулиращите прокоагулантни фактори и активиране на тромбоцитите.
В исторически план при пациенти с ИПАХ са описани три характерни хистопатологични промени: плексогенна белодробна артериопатия, тромботична белодробна артериопатия и белодробна венооклузивна болест. През последните години белодробната капилярна хемангиомиоматоза бе прибавена като отделна характеристика. Pietra et al. класифицират белодробните васкуларни болести, разделяйки първичната белодробна артериопатия на пет отделни субкласа в зависимост от структурните аномалии, обхващащи артериалната стена и лумена (Табл. 1)22.
Таблица 1. Хистопатологични промени при ИПАХ (по Pietra et al.).

Тъй като плексиформените лезии отразяват тежестта на хипертонията, някои субтипове на първична белодробна артериопатия могат да се представят със случаи, при които в биопсичния материал липсват или все още не са развити плексиформени лезии. Освен това последните често са локални и разпокъсани, поради което могат и да липсват. Плексогенната белодробна артериопатия се характеризира с увеличаване на броя на гладкомускулните клетки в артериалното съдово дърво, резултиращо в хипертрофия на медията, експанзия на мускулния слой към лумена на артериолите, което води до т.нар. мускуларизация и може да има за последствие фиброза на интимата и облитерация на съдовия лумен. Всеобща находка е наличие на тромбози в малките артерии, обаче те се считат за вторичен феномен. Плексогенната форма се среща при 28-80% от всички случаи на ИПАХ23, 24.
Тромботичната артериопатия се среща при 40-56% от пациентите с ИПАХ и се характеризира с ексцентрична интимална фиброза и данни за реканализирани тромби in situ. Налице са разпростаранени тромбози на малките съдове, комбинирани с белези на средна белодробна хипертензия (хипертрофия на медията, мускуларизация, интимална пролиферация).
Венооклузивната болест се среща в по-малко от 7-16% от случаите на ИПАХ и се характеризира с интимална пролиферация и фиброза на интрапулмоналните вени и венули.
Капилярната хемангиомиоматоза се характеризира с интерстициална пролиферация на тънкостенното капилярно съдово легло, резултиращо в хипертензия на околните структури и компресия на интрапулмоналните венули.
Патофизиология
Патофизиологията на ИПАХ не е изяснена добре. Обсъжда се тезата за атака на ендотела на белодробните съдове от хормонални, механични и други фактори, водещи до повишена възприемчивост и белодробно съдово увреждане (т.нар. теория за множествените увреждания). По-нататък резултатът е каскада от събития, характеризиращи се със засягане на съдовете, ендотелна дисфункция и гладкомускулна пролиферация на интимата и медията. Основните патогенетични механизми на ИПАХ обаче все още не са достатъчно добре изяснени, много от тях са в сферата на предположенията. В най-общия случай обаче е необходимо наличие на индивидуална (вероятно генетична) възприемчивост и наличие на различен брой стимули, водещи до идентични промени на съдово увреждане и възстановяване (белодробно съдово ремоделиране) и оттук – до ПХ.
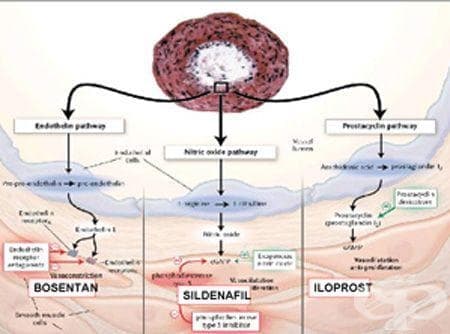
Вазоконстрикцията и пролиферацията на гладкомускулни клетки в белодробните артерии са важна особеност в патогенезата на ИПАХ. Ендотелните клетки вероятно играят важна част от процеса на ремоделиране25-29. Мутации на рецептора за TGF-β могат да доведат до моноклонална ендотелна клетъчна пролиферация и до нарушена регулация на растежа на ендотелните клетки, имаща за резултат плексиформени лезии26. Възможната роля на ендотелните клетки в патогенезата на ИПАХ поставя въпроса дали това е изолирана белодробна болест, или е част от системна такава. До този момент липсват преки доказателства за това. Вазоактивните медиатори вероятно играят важна роля за ПАХ25, 26, 27, 29. Увреждането на ендотелните клетки причинява дисбаланс между вазодилататори (азотен окис, простациклин) и вазоконстриктори (ендотелин-1, тромбоксан) и може да доведе до вазоконстрикция и хипертрофия на медията.
Белодробната циркулация е важно място за продукцията и клирънса на ендотелин (мощен вазоконстриктор и гладкомускулен митоген), като активирането на ендотелиновата система се свързва с ПХ. Ендотелинови рецептори тип А са открити в белодробните гладкомускулни клетки, медиирайки контракция на гладката мускулатура, пролиферация на гладкомускулни клетки и фибробластите. Ендотелиновите рецептори тип В са локализирани както върху ендотелните, така и върху гладкомускулните клетки, медиирайки вазореактивност и вземайки участие в повишаването на съдовия тонус и белодробната съдова хипертрофия.
Серотонинът е белодробен вазоконстриктор, който съдейства за растежа на съдовете и е повишен при пациенти с ИПАХ29. С това може да се обясни по какъв начин fenfluramine и dexfenfluramine (инхибитори на серотониновото захващане) могат да се окажат отключващи фактори за ИПАХ при възприемчиви индивиди2.
Алфа-адренергичните рецептори могат също да играят роля в патогенезата на ИПАХ30. Алфа- и β-адренергичните рецептори имат функция при регулиране на белодробния съдов тонус, водейки до вазоконстрикция или вазодилатация. Интрацелуларният калций е друг важен регулатор на гладкомускулната контракция и пролиферация. Стимулирането на β-адренергичните рецептори повишава свободния интрацелуларен калций и по този начин води до вазоконстрикция. Калиевите йонни канали също имат важна роля в определянето на концентрацията на свободния интрацелуларен калций. При пациенти с ИПАХ калиевите канали могат да бъдат блокирани при стимулиране на β-адренергичните рецептори, или пък поради дисфункция могат да доведат до повишаване на интрацелуларния Ca2+, което води до вазоконстрикция31.
Клинична картина
Средното време от поява на симптомите до поставяне на диагнозата се изчислява на около две години. Въпреки днешния стремеж за ранно откриване и своевременно поставяне на диагноза, времето на забавяне не се е променило значимо през последните години. Причина за това е фактът, че ранните симптоми за ИПАХ са неспецифични. Неспецифичното клинично представяне заедно с ниската честота на заболяването правят диагнозата трудна и водят до значително забавяне.
Най-чести симптоми, установени при пациенти с ИПАХ, са диспнея и умора1, 16, 31. Други оплаквания са синкоп, гръдна болка, палпитации, периферни отоци. В много случаи ситуацията е усложнена и от факта, че диагностицирането изисква усилена и пространна дейност за идентифициране и изключването на друга позната причина, водеща до повишаване на налягането в БА. Честотата на представените при ИПАХ симптоми е както следва: диспнея – при 60%, понижен физически капацитет – при 19%, повтарящи се синкопи – при 13%. Всички те по-често се проявяват при жени, отколкото при мъже.
Физикално изследване
При пациенти с ПАХ могат да открият различни отклонения от нормата. Клиничното изследване на сърдечносъдовата система може често да разкрие промени. При аускултация пулмоналната компонента на втори сърдечен тон обичайно е усилена, което може се демонстрира като фиксиран или парадоксално раздвоен тон, представящ се при наличие на тежка деснокамерна дисфункция. Аускултират се патологични деснокамернни III и/или IV сърдечен тон, шум на трикуспидална регургитация. При авансиране на болестта може да се установят периферна цианоза и отоци1,16,18,19. Може да е налице шум на пулмонална (шум на Graham Steell) и на трикуспидална регургитация. Като белег на обемно натоварване или дяснокамерна недостатъчност може да се наблюдава югуларен венозен пулс. Други прояви на ПАХ могат да бъдат хепатомегалия с палпиращи се пулсации върху черен дроб и абнормен хепато-югуларен рефлекс. Асцит се наблюдава при пациенти, които не са лекувани или при такива с влошаване и декомпенсирана десностранна сърдечна недостатъчност. Физикалното изследване на белите дробове обичайно не показва отклонения от нормата. Обективното изследване на крайниците може да установи наличие на различен по степен оток. При пациенти, които принудително спазват леглови режим, обичайно се установяват отоци.
Диагноза
Спорадичната ИПАХ е трудно установима болест до момента на поставяне на диагнозата. Феномен на Raynaud може да се наблюдава при около 10% от пациентите, главно при жени1. За изключване на вторична ПАХ е необходимо да се изследват антинуклеарни антитела, антинеутрофилни цитоплазмени антитела, ревматоиден фактор, тест за HIV-инфекция, чернодробни тестове1, 16, 19, 20. Voelkel et al. докладват за позитивна корелация между повишените стойности на пикочна киселина и нивото на повишаване на налягането в дясно предсърдие32.
Лабораторни изследвания
Антинуклеарни антитела (АНА)
При съмнение за наличие на ИПАХ важен момент е изключването на автоимунни нарушения. Различни проучвания показват, че при около 40% са налице позитивни АНА, без наличие на друга манифестация на автоимунна болест. По-голяма част от автоимунните нарушения обаче, свързани с ПАХ, се дигностицират въз основа на клинични резултати, като серологичните изследвания са допълнително потвърждение на диагнозата.
Тиреоид-стимулиращ хормон
Скринингът за наличие на абнормна функция на щитовидната жлеза при поставяне на диагнозата ИПАХ е необходим, защото пациенти с абнормна функция демонстрират сходни симптоми с ИПАХ. Сам по себе си хипертиреоидизмът може да доведе до повишено налягане в БА.
HIV-инфекция. При HIV-серопозитивни пациенти честотата на ИПАХ е по-висока от тази в общата популация, поради което извършване на тест за HIV е част от рутинната оценка на пациентите.
Образни изследвания
Торакална рентгенография
Рентгеновото изследване на гръдната клетка може да бъде първа диагностична стъпка за диагнозата при пациенти с диспнея, обаче при много пациенти с ПАХ резултатите не дават възможност за поставяне на етиологична диагноза. Рентгенографията дава възможност да се изключат други причини за ПАХ, като интерстициални и алвеоларни нарушения, които могат да доведат до медиирана от хипоксия белодробна вазоконстрикция. Това изследване показва повишен кардиоторакален индекс, проминиране на белодробните артерии, разширяване на хилусните съдове и редукция на периферните белодробни съдове. При 6% от пациентите с ИПАХ обаче рентгенографията не показва отклонения от нормата1, 19.
Ехокардиография (Ехо-КГ)
Трансторакалната Ехо-КГ е ценен инструментален метод за диагнозата на ИПАХ с оглед преценка на левокамерната и деснокамерната функция, определянето на белодробното систолно артериално налягане, както и изключването на конгенитални аномалии и клапни заболявания. Резултатите от Ехо-КГ могат да демонстрират наличие на шънт през foramen ovale, което се среща при 33% от пациентите. Тя показва дилатация на десните сърдечни кухини, деснокамерна хипертрофия или повишаване на скоростта на регургитация през трикуспидалната клапа.
Високоразделителна компютърна томография (HRCT) и вентилаторно-перфузионна сцинтиграфия (VQ scan). Тези два образни метода се използват най-често, за да изключат наличието на интерстициална белодробна болест и/или белодробен тромбоемболизъм (БТЕ). Перфузионната сцинтиграфия може да бъде нормална или да покаже отклонения от нормата, като демонстрира неравномерни дифузни нарушения в съответствие с васкуларната обструкция. Вентилаторната сцинтиграфия не показва отклонения от нормата1, 16.
Белодробна ангиография
Това изследване обичайно се извършва с оглед диференциална диагноза на налична БТЕ. В същото време обсъждането на тази високорискова процедура при пациенти с повишено белодробно артериално налягане и/или деснокамерна недостатъчност трябва да бъде добре обмислено. Дифинитивната диагноза се поставя след извършването на дясна сърдечна катетеризация: установяват се повишено СПАН, повишено налягане в дясно предсърдие (ДП), нормално капилярно налягане и средно понижен сърдечен индекс (СИ)1. С помощта на това изследване се отхвърля възможността за наличие на интракардиален шънт. По време на осъществяването на дясна сърдечна катетеризация се извършва и вазореактивен тест; позитивен отговор се приема при спадане на СПАН и повишаване на СИ, което е добър прогностичен белег.
Други изследвания
Електрокардиография(ЕКГ)
Най-общо показва белези на десностранно сърдечно обременяване – изместване на сърдечната ос надясно, деснокамерна хипертрофия и дилатация1, 19. При някои от пациентите са налице само част от тези промени, при някои липсват промени и в този смисъл нормалната ЕКГ не изключва наличие на ИПХ.
Белодробна функция и кардиопулмонален тест с натоварване
Функционалното изследване на дишането и оценката на вентилаторната функция са от помощ за диференциране на белодробните съдови заболявания от наличните сърдечни нарушения, както и на рестриктивните от обструктивни белодробни заболявания.
Функционалното изследване на дишането (ФИД) при пациенти с ИПАХ обичайно показва леко изразен рестриктивен синдром на вентилаторна недостатъчност, без наличие на обструктивно нарушение1, 18, 19. Дифузионният капацитет обичайно е редуциран около 70% от предвидения1.
Кръвно-газов анализ (КГА). При ИПАХ показва най-често наличие на хипоксемия и хипокапния1, 17, 18. При болни с ПАХ е увеличена P(A-a)O2 и PaO2 е понижено. При пациенти с лека до средно тежка левостранна сърдечна недостатъчност тези стойности са нормални.
Шестминутен кардиопулмонален тест с ходене. Показва обратна корелация с хемодинамичните параметри и се явява независим предиктор за преживяемост. При пациенти с ИПАХ нивото на върховата кислородна консумация по време на натоварването, кислородният пулс и вентилаторните еквиваленти са абнормни и варират в различна степен.
Изследване на съня. При наличие на клинични индикаци при пациенти с вероятна ИПАХ трябва да бъде осъществено изследване на съня, понеже наличието на сънна апнея се явява причина за ПАХ.
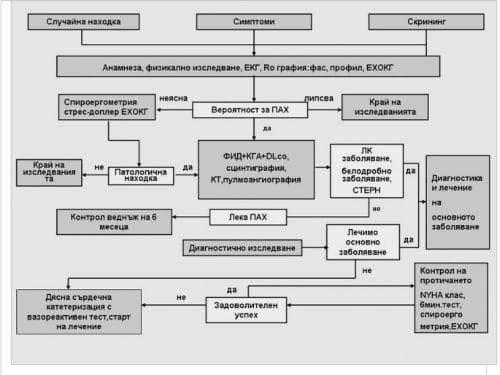
Диагностични алгоритми. В литературата са известни редица диагностични алгоритми, предложени за подпомагане на преценката и изключване на причини, водещи до развитието на вторична ПАХ (Фиг. 1). Основната препоръка при всички тях е следната: при наличие на триада от диспнея, „нормална” рентгенография и нормални стойности на ФИД да се мисли за ПАХ.
Фигура 1. Диагностичен алгоритъм за ИПАХ, модификация (по Olschewski et al.).
Стадиране
За определяне тежестта на болестта обичайно се използва добре познатата класификация на New York Heart Association/World Health Organization (NYHA) за определяне на функционалния клас. Обаче тази система очевидно има ограничено приложение, тъй като е субективна. Други опити за характеризиране на тежестта включват оценка на хемодинамичните промени, след осъществяване на дясна сърдечна катетеризация, оценка на физическия капацитет (върхова О2-консумация при теста с натоварване, 6-минутен тест с ходене) и клинична оценка на тежестта на сърдечната недостатъчност, включваща симптоми и отклонения от нормата в хода на обективното изследване. Позитивни резултати за серумен тропонин и наличие на перикарден излив имат също така прогностично значение, предсказващо по-лоша прогноза.
Диференциална диагноза. ИПАХ трябва да бъде разграничена от хронично белодробно сърце, дилатативна кардиомиопатия, хипотиреоидизъм, митрална стеноза, смесена съединителнотъканна болест, портална хипертония, кардиогенен белодробен оток, БТЕ, вторична пулмонална хипертония, стеноза на белодробната артерия, склеродермия, системен еритематоден лупус.
Лечение
По традиция се характеризира със сложност и малък брой терапевтични възможности98. В тази област през последните години бяха направени големи усилия. От една страна, това се дължи на новите разбирания за патофизиологичните механизми на белодробните съдови заболявания, а от друга страна, на нарастващия брой на нови медикаменти. Днес е почти сигурно, че една от основните аномалии при тази болест е дисфункцията на ендотела, която резултира в понижена продукция на вазодилататорни субстанции като NO или простациклин, както и в повишена продукция на вазоконстрикторни субстанции като ендотелин или тромбоксан. Разбирането на тези механизми даде възможност за промяна на насоките в терапията, която от една чисто вазодилататорна терапия с калциеви антагонисти премина към поведение, имащо за цел да промени ремоделирането на белодробното съдово легло. Това доведе до революция в прогнозата на заболяването. Резултати от рандомизирани и контролирани проучвания при възрастни показват, че някои терапевтични схеми на лечение могат да променят протичането на болеста. Днес е възможно да се подобрят не само симптомите и качеството на живот, но и преживяемостта на пациентите с ИПАХ.
Контрол на протичането
Пациентите с тежка ПАХ се нуждаят от непрекъснат контрол в специализиран център. Интервалите на проследяване трябва да се планират за пероид от 3 ± 6 месеца. Параметрите за контрол, които имат прогностично значение, са функционалният клас по NYHA и 6-минутният тест с ходене, определени ехокардиографски и спиро-ергометрични показатели. Неблагоприятен прогностичен белег са анамнестичните епизоди на десностранна сърдечна декомпенсация. Определянето на натриуретичните пептиди би могло да придобие роля на важен прогностичен маркер. Ехокардиографското определяне на систолното налягането в БА има ограничено значение за протичане на заболяването. Повторна сърдечна катетеризация е уместна при вземане на важни решения за продължаване на лечението, като например промяня в терапевтичната схема или решение за трансплантация.
Диана Петкова, д-р, гл. асистент, Клиника по пневмология и фтизиатрия, МБАЛ „Св. Марина” – Варна, Медицински университет – Варна
Литература
1. Galie N, Torbicki A, Barst R et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J 2004; 25 (24): 2243-2278
2. McDonnell PJ, Toye PA, Hutchins GM. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis 1983;127:437-41
3. Lebrec D, Capron JP, Dhumeaux D, Benhamou JP. Pulmonary hypertension complicating portal hypertension. Am Rev Respir Dis 1979; 120: 849-56
4. Edwards BS, Weir EK, Edwards WD, Ludwig J, Dykoski RK, Edwards JE. Coexistent pulmonary and portal hypertension: morphologic and clinical features. J Am Coll Cardiol 1987; 10: 1233-8
5. Petitpretz P, Brenot F, Azarian R, et al. Pulmonary hypertension in patients with human immunodeficiency virus infection. Comparison with primary pulmonary hypertension. Circulation 1994; 89: 2722-7
6. Mesa RA, Edell ES, Dunn WF, Edwards WD. Human immunodeficiency virus infection and pulmonary hypertension: two new cases and a review of 86 reported cases. Mayo Clin Proc 1998; 73: 37-45
7. Golpe R, Fernandez-Infante B, Fernandez-Rozas S. Primary pulmonary hypertension associated with human immunodeficiency virus infection. Postgrad Med J 1998; 74: 400-4
8. Gurtner HP. Aminorex and pulmonary hypertension. A review. Cor Vasa 1985; 27: 160-71
9. Brenot F, Herve P, Petitpretz P, Parent F, Duroux P, Simonneau G. Primary pulmonary hypertension and fenfluramine use. Br Heart J 1993; 70: 537-41
10. Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. N Engl J Med 1996; 335: 609-16
11. Dawkins KD, Burke CM, Billingham ME, Jamieson SW. Primary pulmonary hypertension and pregnancy. Chest 1986; 89: 383-8
12. Kleiger RE, Boxer M, Ingham RE, Harrison DC. Pulmonary hypertension in patients using oral contraceptives. A report of six cases. Chest 1976; 69: 143-7
13. Collins E, Hardwick RJ, Jeffery H. Perinatal cocaine intoxication. Med J Aust 1989; 150: 331-2
14. Gomez-Sanchez MA, Saenz de la Calzada C, Gomez-Pajuelo C, Martinez-Tello FJ, Mestre de Juan MJ, James TN. Clinical and pathologic manifestations of pulmonary vascular disease in the toxic oil syndrome. J Am Coll Cardiol 1991; 18: 1539-45
16. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987; 107: 216-23
17. Rubin LJ. Primary pulmonary hypertension. N Engl J Med 1997; 336: 111-7
18. Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet 1998; 352: 719-25
19. Rich S. Primary pulmonary hypertension. Prog Cardiovasc Dis 1988; 31: 205-38
20. Sandoval J, Bauerle O, Palomar A, et al. Survival in primary pulmonary hypertension. Validation of a prognostic equation. Circulation 1994; 89: 1733-44
21. Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, causing familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet 2000; 26: 81-4
22. Pietra GG, Edwards WD, Kay M, et al. Histopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, Primary Pulmonary Hypertension Registry. Circulation 1989; 80: 1198-206
23. Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 1994; 144: 275-85
24. Bjornsson J, Edwards WD. Primary pulmonary hypertension: a histopathologic study of 80 cases. Mayo Clin Proc 1985; 60: 16-25
25. Humbert M, Morrell NW, Archer SL, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43(12 Suppl S): 13S–24S
26. Giaid A, Yanagisawa M, Langleben D, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med 1993; 328: 1732-9
27. Langleben D, DeMarchie M, Laporta D, et al. Endothelin-1 in acute lung injury and the adult respiratory distress syndrome. Am Rev Respir Dis 1993; 148(6 Pt 1): 1646-50
28. Davie N, Haleen SJ, Upton PD, et al. ET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am J Respir Crit Care Med 2002; 165: 398-405
29. Herve P, Launay JM, Scrobohaci ML, et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med 1995; 99: 249-54
30. Salvi SS. Alpha1-adrenergic hypothesis for pulmonary hypertension. Chest 1999; 115: 1708-19
31. Takizawa T, Hara Y, Saito T, et al. Alpha1-adrenoreceptor stimulation partially inhibits ATP sensitive K+ current in guinea pigs ventricular cells: attenuation of the action potential shortening induced by hypoxia and K+ channel openers. J Cardiovasc Pharmacol 1996; 28: 799-808
32.Voelkel MA, Wynne KM, Badesch DB, et al. Hyperuricemia in severe pulmonary hypertension. Chest 2000; 117: 19-24
Коментари към Идиопатична пулмонална артериална хипертония